|
|
DNA Sequence Assembler
Your first script
To create your first script, create a new empty file, name it something like SeqAssembly.cmd and paste the code in the examples below in it.
Parameters
The parameters can be provided with -- or / Example: --InputFolder /InputFolder
When you provide paths it is not mandatory to put them in double quotes: Example: Accepted: --InputFolder="C:\My Samples" Also accepted: --InputFolder=C:\My Samples
Minimal example
The only mandatory paramater you need to specify is 'InputFolder'.
Classic example
The script above will call DNA Baser Console using 'c:\samples' as input folder. All ABI samples/chromatograms in that folder will be assembled. The DNA Baser Console exactly as DNA Sequence Assembler: it will automatically import samples, automatically trim low quality ends, remove recognitions sequences, correct the ambiguities and save the log and the contig to disk (in the c:\samples\out' folder in this example).
Run sequence assembly at batch
If you want to assemble all sequences in 1000 folders you can do something like this:
Or better, you can use a FOR loop as shown here:
More details about the FOR loop here: https://stackoverflow.com/questions/138497/iterate-all-files-in-a-directory-using-a-for-loop
Help
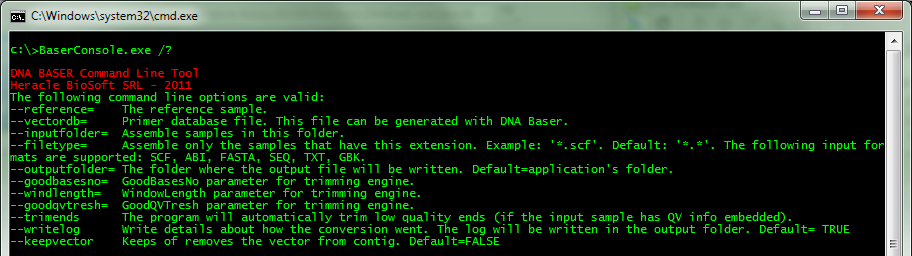
At the console, type BaserConsole.exe /? to get help:
--inputfolder=path Assemble samples in this folder --outputfolder=path Optional. The folder where the output file will be written (default=input folder) --reference=file Optional. The reference sample. Make sure it is located in a folder different than the input folder --filetype=string Optional. Assemble only the samples that have this extension. Example: '*.scf'. The following input formats are supported: SCF, ABI, FASTA, SEQ, TXT, GBK. (default: *.*) --vectordb=file Optional. Primer database file. This file can be generated with DNA Baser --writelog=boolean Optional. Write details about how the conversion went. The log will be written in the output folder (default: true) --keepvector=boolean Optional. Keeps of removes the vector from contig. (default=FALSE) --trimends Optional. The program will automatically trim low quality ends (if the input sample has QV info embedded) (default: true) --goodbasesno=int Optional. GoodBasesNo parameter for trimming engine (default: 60) --windlength=int Optional. WindowLength parameter for trimming engine (default: 16) --goodqvtresh=int Optional. GoodQVTresh parameter for trimming engine (default: 20) --wordsize=int Optional parameter for contig assembler. (default: 18) --identproc=int Optional parameter for contig assembler. (default: 65) --minoverlap=int Optional parameter for contig assembler. More details on our web site. (default: 20)
Known issues
The InputFolder and OutputFolder should not end with a backslash character (\). Example: Accepted: --InputFolder=C:\MySamples NotAccepted: --InputFolder=C:\MySamples\
|
||||||