| |
|
|
|
|
|
Sequence assembly FAQ
DNA Baser shows the correct contig on screen but when I save it to disk it is incomplete
This is because you Vector Removal settings are wrong. You need to use different vector recognition sequence, which after RC (reverse complement) are not identical. Please see this page for details.
Why DNA Baser cannot assemble (some of) my files?
A quick look into the LOG will tell you why. In most cases is because DNA Sequence Assembler automatically removed some of your samples from the job list. This happens when the quality of the samples is below a certain threshold. To force DNA Baser to keep even the low quality samples, you need to relax the “Trimming engine” parameters (DNA Baser can automatically trim low quality ends of your samples) and the “Assembler engine” parameters. However, this may result sometimes in more errors/ambiguities in your contig which means more manual work to correct them. Therefore, if you have enough redundant samples in the set of files that you want to assemble, it is better to let DNA Baser to exclude the samples with low quality (the samples that may introduce ambiguities). Another reason may be that the sample does not really belong to the current contig. In this case you will get a message saying "Sample [xxx] has no neighbors".
DNA Baser does not create the contig with the right orientation
The SCF/ABI/FASTA samples you receive from your sequencing machine are in random order (each user can use its own naming scheme). Therefore, DNA Baser cannot know the orientation of your contig. We will add in the near future an input box where you can designate on of your samples as being forward or reverse. The orientation of the contig will then be changed according to this information. Workaround:
Do I have to save the contig every time I assemble some samples?
I have an error saying: Cannot open file… The process cannot access the file because it is being used by another process” or “EFOpenError".
DNA Baser does not lock the files that it opens. This way it allows you to open the same file multiple times without generation sharing violations.
DNA Baser does not cut the recognition sequence (vector) correctly OR DNA Baser saves only a small part (only the vector) of the contig to disk
This happens when the two recognition sequences used are the exact reverse complement of each other. To solve this, you need to change one of the recognition sequence (e.g. by adding more bases) such that, when reverse complemented, it will not be identical with the other recognition sequence.
I need a small feature. Can you implement it?
How to copy/print chromatograms from DNA Baser?
This can be achieved by copying the chromatograms to clipboard then pasting them in your preferred image editor (example: Corel, Photoshop, Paint Shop Pro, PaintBrush) or text editor (example: Microsoft Word, Wordpad, Open Office). From these editors you can edit or print the image (screenshot) easily. To copy the chromatogram to clipboard click the chromatogram you want to copy then:
Note: new features for chromatogram manipulation has been added in DNA Baser v4.0
How much RAM does DNA Baser Assembler need?
For small contig assembly 1GB of RAM should be more than enough. Please see this page for details about memory requirements.
FAQ about file formats
What file formats does DNA Baser Assembler recognize?
Why I should prefer chromatogram files (SCF/ABI) over Fasta files?
DNA Sequence Assembler is a tool like no other. One of its outstanding features is the capability to perform time consuming task (task that until now could only be done manually) instantly. For example by using DNA Sequence Assembler you do not have to inspect your contig or manually correct the ambiguities. DNA Sequence Assembler will do it for you. However, this feature works only if your input samples contain information about base quality (aka QV or confidence score or basecaller confidence). Therefore, whenever possible you should use chromatogram files (SCF/ABI) files instead of FASTA files, empowering DNA Baser to automatically remove untrusted regions and to automatically correct the ambiguities. The accuracy of the suggestions can be as high as 95-99% and in most cases you don't have to do any manual editing/corrections in your contig. This will save a lot of your precious time!!
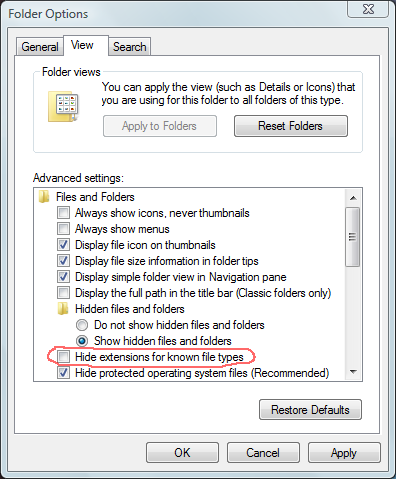
How can I change the file extension of a file (sample)?
|
|||
| SciVance Technologies |