|
|
 |
NextGen Workbench The way the bioinformatics should be...
How to convert SFF to FastQ files
The following conversions are possible:
How to convert a SFF file to FastQ in NextGen Workbench v2?
NextGen Workbench v2 has a dedicated SFF to FastQ converter. This is much much faster than the converter offered in v1.
How to convert a SFF file to FastQ in NextGen Workbench v1?
This short tutorial will show you how to use NextGen Workbench to convert your SFF files to FastQ.
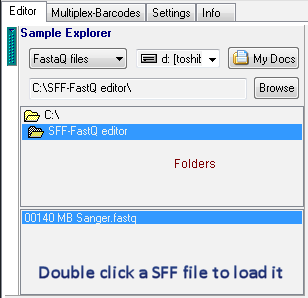
1. In Sample Explorer, double click a SFF file to load it:
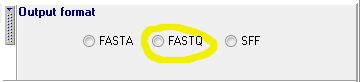
2. Set the output format to 'FastQ':
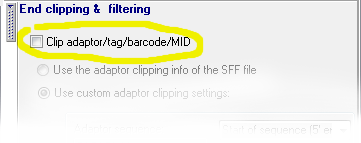
3. Optional step: If you want to perform additional processing (for example to clip adaptors) check the appropriate box:
4. Press the 'Refresh and save' button to save the file. NextGen Workbench will ask you where to save the file.
Converting multiple SFF files at once
You can start multiple sessions of NextGen Workbench if you want to convert multiple files at once. You can open the SFF/FastQ files by double clicking them in Windows Explorer after you associate NextGen Workbench with SFF/FastQ files.
About bottlenecks
We don't recommend to start more sessions than the number of CPU cores you have. You won't see additional speed improvement after you match the number of CPUs. So, if you have a 16 core CPU you can start up to 16 instances. However, the limit may be much however if you have a slow hard drive. The program needs to read AND write a lot of data. SFF/FastQ are usually pretty large. So your CPU core will not be fully used if your hard drive cannot keep up (read and write data fast enough).
Notes about SFF to FastQ conversion
All these are format incompatibility not a limitation in NextGen Workbench.
|
||